
Introduction
Throughout the history of medicine, one of the most relevant discoveries has been that of deoxyribonucleic acid (DNA) by Watson and Crick in 1953. This finding marked a before and after in human genetics; so much so that two historical periods are described: a classical era and a new genetics [1].
Classical genetics is distinguished by being the great era of clinical observation, through which phenotypes were established in patients in order to recognize certain characteristics in physical examination and thus guide a diagnosis [2]. In addition, new terms were introduced to the medical community, such as eugenics, which aims to intentionally manipulate the genome to eliminate defects in it.
This brought repercussions and new discussions about ethics and human dignity, questioning the human value by marginalizing and segregating those who, by natural chance, were different. However, up to this point in the history of humanity and medicine, the mechanisms of genetic expression were not deeply understood, which is why many schools of thought adopt extreme positions regarding how humans should be phenotypically [3].
On the other hand, the New Genetics is considered the great era of the human genome, distinguished by elucidating the mechanisms of gene expression and the genes that encode DNA. This has allowed the development of projects such as the mapping of the human genome, promoting progress in genetic understanding and facilitating the study of hereditary and complex diseases.
Furthermore, it has made personalized treatments possible and the beginning of gene therapy. Likewise, it has led to the use of new molecular biology techniques such as Polymerase Chain Reaction (PCR), developed in 1983 by Karry Mullis, which has been fundamental for clinical research by detecting small nitrogenous bases in specific DNA sequences. Equally important have been the techniques of restriction enzymes and molecular hybridization [4].
Thanks to these advances throughout history, a countless number of knowledge has emerged that has allowed the evaluation of clinical phenomena at a molecular level. The purpose of this literature review is to keep the scientific community updated on the different genetic pathologies and their classification taking into account the nomenclature and key points of each [3].
Genetic Pathologies
Genetic Pathologies are classified into three groups according to Thompson & Thompson [2] and Dr. Pilar Carvallo [5], which are:
1. Genopathies or Monogenic Disorders: only one gene involved in the disease per patient.
2. Chromosomopathies or Chromosomal Disorders: refers to alterations in the number of chromosomes or to chromosomal translocations.
3. Multifactorial Disorders: there is more than one gene involved in the disease, in addition to environmental factors.
Therefore, monogenic disorders are subcategorized into [5]:
a. Autosomal Dominant: it has 3 main characteristics:
I. All affected individuals have an affected parent.
II. Only one of the two alleles for the same gene needs to be altered for the disease to occur.
III. Both sexes are affected in similar proportions.
b. Autosomal Recessive: its three main characteristics are [5]:
I. There is usually no previous family history.
II. The highest probability of finding a second affected individual is a sibling of the patient under study.
III. Both alleles of the same gene must be altered for the disease to occur. For that reason, both parents must be carriers of the alteration in one of their alleles (Increased probability in consanguineous marriages).
c. X-linked: can be dominant or recessive. This type of inheritance is associated with genes found on the X chromosome. Since women have two X chromosomes and men only have one, the way this chromosome is inherited affects males and females differently, predominantly in males [5].
Other authors like Ángel Herraez [6], classify them as:
1. Disorders according to the type of cell (Germinal or Somatic).
2. Disorders according to magnitude (Large mutations, Medium mutations, and Point mutations).
3. According to the mechanism: Endogenous mutations (Errors in replication, Oxidative spread of bases, Endogenous mutagens) and Exogenous mutations (physical agents, chemical agents, or biological agents).
But for the purposes of this work, we will maintain the classic classification mentioned at the beginning.

Genopathies
Genetic diseases occur when there is an alteration in a DNA gene, whose proper functioning is necessary for the normal development of an individual. In this way, when changes occur in the quantity and/or function of genes, mutations or genetic variations occur, which differ depending on the type of altered sequence and therefore, the severity of them [7].
These genopathies have an identifiable component that is responsible for their variation and in order to identify it, the study methods have advanced in such a way that they allow analyzing the genome structure to issue the diagnosis at the molecular level [8]. These phenomena occur in principle at the molecular level and are observable.
A mutation implies molecular changes that do not exceed 10 megabases (MB), changes that exceed this megabase value are chromosomal anomalies (which are observable) [8].
To objectify the genomic findings, authors have varied in their definitions, with some classifying them as chromosomal [9], and under the same concept [10], subclassifying them into:
- Suppression mutations.
- Insertion mutations.
- Inversion mutations (pericentric or paracentric).
- Transposition mutations (referring to translocations within the same chromosome).
- Reciprocal translocation mutations.
- Polysomy mutations.
- Mutations by aneuploidies.
However, this classification has several points in which it resembles the classification of chromosomal aberrations, therefore, as a key point of this monograph, we will maintain the original concept of mutations by molecular changes [9].
Mutations can be classified according to their structure, function, cause of origin, cell in which it occurs, from an evolutionary point of view, the extent of the genetic change, and molecular pathology.
From the perspective of Molecular Pathology, mutations are classified as [9,11]: Point mutations, Transposition mutations, Splacing failure mutations, deletion or insertion mutations, Gene fusion, Triplet repeat (See table 2).
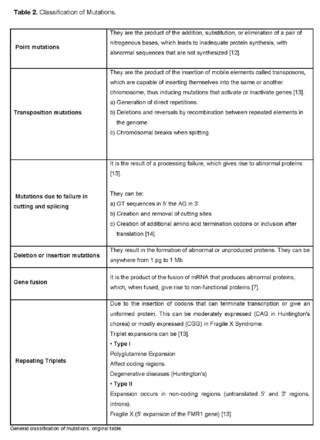
In order for these genomic changes to occur, there must be certain effects on the function of proteins and any of the following steps can be altered: transcription, translation, polypeptide folding, post-translational modification, assembly of monomers into a polymeric protein, subcellular localization of the polypeptide or holomer, cofactor or prosthetic group that binds to the polypeptide, function of a properly folded, assembled and localized protein produced in normal amounts [11].
The consequences of mutations can result in alterations at the level of enzymes as occurs in Inborn or Congenital Errors of Metabolism or defects in structural proteins such as:
- Intracellular - > intermediate filaments - keratins
- Extracellular - > collagen
- Formers of nuclear receptors and channels (RFTQ)
- Transporters - > hemoglobin
Chromosomal abnormalities
Since 1956, when innovative techniques for the study of chromosomes were developed, disorders associated with chromosomal information alteration have been evidenced [15,16]. Turnpenny, Ellard, and Cleaver classify chromosomal aberrations into numerical, structural, and mixoploidies chromosomopathies, which will be discussed later [4].
Structural chromosomal aberrations
These alterations are the result of disordered union following chromosome breakage [17]. This alteration can be balanced when the chromosomal information does not undergo loss or gain of genetic information, making them mostly harmless alterations in most cases [18]. On the contrary, unbalanced structural chromosomal aberrations imply a significant alteration in the genetic material and therefore major clinical implications [4,17].
Regarding the classification of structural chromosomal alterations, several sub-classifications have emerged, among which we can find the classification by Yilmaz, Beck, and Lee in the book "Genetics and Genomics in Medicine" where they group the different alterations into categories, as follows [18]:
- Abnormal chromosomal segregation: aneuploidy and uniparental disomy.
- Recurrent chromosomal syndromes: duplication, deletion, alteration in copy number.
- Non-recurrent chromosomal anomalies: deletion syndromes, gene disruption.
- Unbalanced familial anomalies: translocations and inversions.
- Syndromes related to genomic imprinting.
Despite this being one of the most recent and complete classifications, there is another classification presented by Turnpenny, Ellard, and Cleaver in 2021, which is as follows [4]:
- Translocations: This type of alteration occurs when there is a break in a chromosome segment and it is transferred to another [19], there are different types that can occur when a translocation occurs, among these variations we find:
- Robertsonian translocation: It is that alteration of two acrocentric chromosomes which merge at the centromere to form a single chromosome [20].
- Reciprocal translocation: It occurs when two different chromosomes break and exchange a segment of similar size between each other [4].
- Deletions: It is a type of mutation that involves the loss of one or more nucleotides from a segment of DNA [21].
- Insertions: Consists of the addition of one or more nucleotides from a segment of DNA, within which includes [22, 23]:
- Investments: Occurs when two breaks of two different segments occur in the same chromosome and these are inverted [24], there are two types:
- Pericentric mutations: When the inversion involves the centromere.
- Pericentric mutations: When the inversion occurs in the same arm.
- Chromosome in ring: This alteration involves a break in each arm of a chromosome leaving the central ends free, which will form a ring and the distal portions are lost [25].
- Isocromosomes: An isochromosome is identified because it shows the loss of one arm with subsequent duplication of the existing arm, its clinical relevance is because in 15% of cases it results in Turner Syndrome [4].
Number aberrations
Genetic alterations in relation to the number of chromosomes must be remembered that the normal number of human chromosomes is 23 pairs, and this number may vary in relation to the genetic alterations that occur, numerical chromosomal aberrations have had multiple classifications throughout history, however, there is no significant difference between each author so we have reached a consensus.
- Aneuploidy: This is the condition where there are more or fewer chromosomes than physiologically expected, and it is due to inadequate chromosomal separation during mitosis or meiosis [15,26].
In the group of aneuploidies there are some well-known trisomies that are related to the neonatal stage, these are trisomy 13, trisomy 18, and trisomy 21, also known as Patau syndrome, Edwards syndrome, and Down syndrome respectively [27], these syndromes are easily identifiable due to their characteristic phenotype, there are other trisomies like trisomy 16, however, they are usually associated with early pregnancy losses [4].
There are also trisomies of the sex chromosomes presenting the triple X syndrome (XXX), Klinefelter syndrome (XXY), Jacob syndrome (XYY) [28] among other less common ones.
Likewise, aneuploidies occur in cases where a chromosome is missing, this presentation is called monosomy, in most cases it tends to be incompatible with life [4], however, among the more frequent presentations is Turner syndrome (X0) which is associated with a high number of early miscarriages but is also compatible with life once conceived [26].
- Polyploidies: Occur when two or more copies of a chromosome are present, which alters the genetic load in multiple ways, often incompatible with life [4,26].
- Mixoploidies: There are two types: mosaicism and chimerism.
Mosaicism refers to chromosomal alterations where two different cell lines with the same zygotic origin are present in a tissue of an individual, indicating the same genetic origin [29]. Conversely, chimerism occurs when two different cell lines with different genetic origins are present in a tissue [18].
In turn, chimeras are classified as follows:
- Dispermias: Occurs when two ova are fertilized by two spermatozoa with different genetic material, consequently, the resulting zygotes will merge to form a chimeric embryo [4,18].
- Blood: It occurs when two non-genetically identical twins exchange cells in utero through the placenta, normally this produces a series of sexual alterations [4].
Multifactorial Disorders
They are the result of the interaction of the genome with the environment, these environmental factors are called teratogenic agents [30], they are the result of the combined effects of multiple genetic variants and environmental risk factors, accompanied by genetic and environmental risk factors that alter susceptibility to the disease [31].
For the reasons mentioned, these disorders are considered to have a multifactorial origin, in this section it is important to emphasize that familial clustering generates an inheritance pattern called "complex" due to the contribution of chance events and the combined effects of multiple environmental and genetic risk factors that are not easy to predict and can lead to a wide variety of disease segregation patterns in families [2].
According to pathogenic criteria, multifactorial disorders can be classified in various ways, for the purposes of this study we will talk about: Associations, Sequences, Disruptions, Dysostosis, Osteochondrodysplasias and Syndromes [31].
1. Associations:
Non-random and statistically significant genetic or environmental factors, in which multiple anomalies are generated, for which a specific etiology has not been described [32].
Also, they can interfere with blastogenetic processes, that is, the events that occur during early embryogenesis, especially during the stages of cell division and formation of germ layers. In this way, when the embryo is a single field of development, defects occur in organs and body segments apparently unrelated to each other, some associations may share a phenotypic overlap in relation to common etiological and pathogenic mechanisms [31].
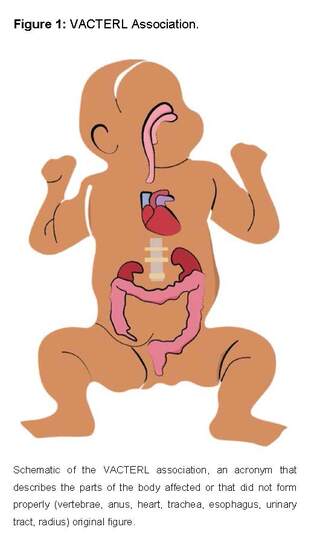
A good example are newborns with the "VACTERLS" association which is characterized by the presence of at least three of the following signs from the acronym in English (vertebral defects, anorectal atresia, cardiac anomalies, tracheoesophageal fistula, esophageal atresia, long-term renal anomalies, limb defects, and single umbilical artery) [33,34].
This association is usually sporadic with a low risk of recurrence, is believed to be multifactorial in etiology, based on a genetically susceptible genome on which environmental triggers act, including teratogens, and is more common in children of diabetic mothers. This is classified as an association, since there is no evidence that the malformations are related from a pathogenetic point of view, but these malformations appear together with an incidence higher than expected by chance [33,34].
2. Sequences:
They are defined as a pattern of multiple anomalies derived from a single anomaly followed by a cascade of side effects [32], they can depend on both genetic and environmental factors and are characterized because multiple organs and systems can be involved concomitantly [31,35].
An example of common sequences observed in the neonatal period is the oligohydramnios sequence or Potter sequence, which is secondary to the presence of malformations or alterations at the level of the genitourinary system, among which we can find bilateral renal agenesis, severe polycystic kidneys, and even obstruction of the urinary tract, which leads to the presence of oligohydramnios resulting in deformities in the face and extremities due to compression, pulmonary hypoplasia, pneumothorax, and/or growth restriction [32,36].

Other examples of sequences include congenital heart defects, esophageal and duodenal atresia, imperforate anus, sirenomelia, hypoplastic nails, and the Pierre Robin sequence [32,37].
3. Interruptions:
It is a structural defect resulting from an extrinsic insult to a process of originally normal development [32].
This occurs due to exogenous factors that act during intrauterine life such as any substance introduced into the human body (drugs, alcohol, tobacco) or produced by maternal metabolism in specific situations, which can cross the placenta, reach the fetus, affect embryofetal development, and thus generate morphogenetic defects in related body regions [31,38].
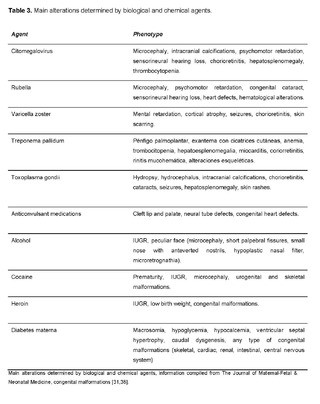
Table 3 exemplifies the main alterations determined by biological and chemical agents:
4. Syndromes:
It is defined as a recognizable pattern of anomalies that is considered to have a specific cause [32], within the syndromes, we find numerical aberrations, which usually have a prezygotic origin; if they arise from postzygotic errors, they are present only in a percentage of cells (mosaics), another group are structural aberrations, which occur de novo from meiotic rearrangements or can be inherited from one of the parents, leading to a balanced chromosomal translocation, as for chromosomal aberrations, it is a subclassification that is discussed in section II of this text [31].
Disostosis:
They are a heterogeneous group of congenital defects with skeletal involvement, due to mutations in genes involved in bone development, since most of the conditions are determined by mutations in fibroblast growth factor (FGFR) genes, on the other hand their classification is based on phenotypic or genetic criteria [39,31].
Dysostoses can be divided into:
- Craniomaxillofacial dysostoses: They are dysostoses that depend on the premature closure of cranial sutures that limits cranial growth, producing a deformation of the skull depending on the affectation of the sutures (type, moment, extension, and symmetry) [31,40].
- Klippel - Feil, a defect in the development of the spine, with possible alterations at the cervical, thoracic, and lumbar levels (short neck, pterygium, kyphosis, and scoliosis); and dysostoses of the limbs, digital defects in one or more adjacent bones [31,40].
6. Dysplasia:
It is an anomaly in the organization or differentiation of cells within a specific type of tissue that results in clinically apparent structural changes [31], in this section, we focus on osteochondrodisplasias, as they are a broad and heterogeneous group of genetically determined conditions that impact the development of bone and cartilaginous tissues [31].
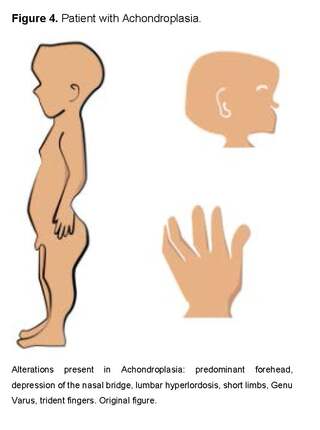
Among the most common osteochondrodysplasias, we find Achondroplasia, which is characterized by megalocephaly, frontal bossing, depressed nasal bridge, hypoplasia of facial bones, prognathism, narrow thorax, rhizomelic short-limb dwarfism, brachydactyly, trident hands and hypotonia, determined by the AD Gly380Arg mutation in the FGFR3 gene, with a high rate of de novo mutation related to advanced paternal age [31].
On the other hand, another case of dysplasia is Osteogenesis Imperfecta, a group of genetically (type 1 collagen genes) and phenotypically (four different clinical variants) heterogeneous conditions, characterized by frequent bone fractures [31].
Malformations
Genetic malformations are defects during morphogenesis. Because their etiology and presentation are very variable, numerous congenital defects have been described, which have been classified considering their etiology, pathogenesis, presentation, and severity [41, 42].
Regarding severity, we must distinguish between a major and minor malformation. The major defect refers to a serious structural anomaly that alters the morphology of an organ and/or tissue, causing a medical or aesthetic pathology that requires medical-surgical treatment in order to restore functionality or in some cases, save the individual's life. On the other hand, minor defects are morphological alterations that affect the individual's quality of life to a lesser extent and do not require life-saving interventions to correct them [41].
In addition, these major defects can be classified to establish the morphological type and the embryonic period in which the alteration manifests and therefore establish its mechanism and predict the variability of the manifestation in individuals (Table 4).
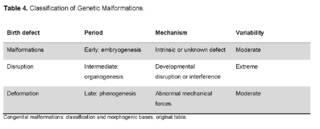
Early defects that occur during embryogenesis are characterized by alterations that involve the entirety of an organ or tissue, preventing its complete or partial development [43,44]. These congenital malformations have a prevalence of 104.5/100,000, being more prevalent in males, in newborns with low birth weight and a gestational age of 37 weeks. It is more common in pregnant women of advanced maternal age.
Regarding mortality, it depends on the type of associated defect, however, the figure can go up to 23.4%, although EUROCAT data estimates the mortality rate as high as 29% [45,46].
Disruptions are organic defects that do not correspond to purely embryological alterations, but rather, due to interferences in the normal development of a tissue, which can be caused by an internal or external agent.
Deformities are structural abnormalities that occur in the fetal period and are due to the interaction of the tissue with abnormal mechanical forces that alter the morphology [47].
Future Perspectives
Research on genetic pathologies is progressing rapidly thanks to current technological advances, facing us with new challenges and unresolved questions, which generates new opportunities for future research [48]. A clear example is CRISPR-Cas9 technology, which was deserving of a Nobel Prize in chemistry in 2020, for opening a significant door that allows the genetic editing of eukaryotic cells [49].
Currently, the CRISPR - Cas9 system is widely used in medicine in areas such as biochemistry, where its ability to block angiogenesis in vitro is being studied [50].
On the other hand, in the field of oncology, it will be focused on the creation of oncolytic viruses [51], and in the treatment of pathologies such as Kaposi's sarcoma [52]. Furthermore, it has been discussed that in the future it will allow for specific modifications in genes with greater precision and lower likelihood of unwanted side effects.
Following the line of gene therapy based on genome modifications, gene therapy using viral and non-viral vectors is based on the administration of nucleic acids using vectors for the treatment of mono and multigenic diseases. The most commonly used viruses have been retroviruses and lentiviruses, and this technique has become popular in the study of diseases such as Hemophilia B, cystic fibrosis, and Parkinson's disease [53].
Similarly, personalized medicine will also play a key role in the research of genetic pathologies, this is due to the widespread use of genomic sequencing techniques, it is increasingly feasible to identify specific genetic variations in each individual, which will allow for designing more personalized treatments that may be more effective and have fewer adverse effects [54].
Furthermore, pharmacogenomics is the area focused on studying the response to drugs and how it is influenced by the genetic variations of individuals, which will be crucial for developing treatments tailored to each patient's genetics and thus maximizing therapeutic outcomes, by predicting the effectiveness and specific doses of treatment according to the patient's genetic characteristics [55].
Finally, the integration of artificial intelligence into genetic research will open an unprecedented door to analyze large volumes of genomic data and identify patterns not perceptible to the human eye [56]. Additionally, these tools could be crucial in identifying new pathogenic mutations and predicting patients' response to different therapies, facilitating the design of more effective and specific treatments for each patient.
Conclusions
Genetic Pathologies are classified according to the level at which they affect the genetic material, at the gene level (genopathies), at the chromosome level (chromosomopathies), and if they are the result of the interaction between the genome and the environment (multifactorial disorders).
Mutations are phenomena that involve molecular changes and depending on which gene is damaged, altered proteins can be produced that generate functional alterations in the body.
Chromosomal anomalies can affect the structure or number of chromosomes and can lead to significant alterations such as deletions, translocations, aneuploidies, and polyploidies which are the cause of different genetic syndromes [1].
Multifactorial disorders are the result of the interaction of teratogenic agents with elements of the maternal genome or the embryo that lead to defects in the formation of tissues and organs [2].
The origin of many malformations remains unknown, but factors such as alcohol or maternal age are important risk factors in their appearance.
Genetic pathologies are a topic of great importance that must be acquired by general practitioners and specialists in any field of medicine, in order to provide an adequate multidisciplinary approach and offer counseling focused on the pathology faced by the medical team.
In Colombia, genetic diseases are a significant cause of infant mortality and it is difficult to understand why they are not included in the Mandatory Health Plan, leaving a significant fraction of the low-income Colombian population without this service, which is why these literature reviews allow us to emphasize on these types of disorders, their complications, and their impact on the quality of life of the patient and their support network, therefore it is of great importance that the health system implement interdisciplinary coverage for the family group and individual undergoing treatment [57,58,59].
In conclusion, future research on genetic diseases will not only seek to find cures for currently incurable diseases, but will also contribute to improving the accuracy and safety of therapies, as well as expanding knowledge about the complex interaction between genes, environment, and disease.