
Genomic variants associated with inborn errors in carbohydrate metabolism in southwestern Colombia
[Variantes genómicas asociadas a errores congénitos en el metabolismo de los carbohidratos en el suroccidente colombiano]Jaime David Viafara Belalcazar1,2,3
 , Jose María Satizabal Soto1,2,3 , Lina Johanna Moreno Giraldo1,2,3
, Jose María Satizabal Soto1,2,3 , Lina Johanna Moreno Giraldo1,2,3 1. Universidad del Valle, Cali, Colombia; 2. Universidad Libre, Cali, Colombia; 3. Grupo de Investigación de Enfermedades Metabólicas Congénitas;
Downloads
Abstract
Introduction: Carbohydrate Inborn Errors of Metabolism (IEMs) result from disruption of the catabolic or anabolic pathways of different carbohydrates, fructose, galactose and glycogen being the most common and belong to a heterogeneous group of disorders that may be inherited or may occur as a result of spontaneous genetic variants. In IEM, there are altered carbohydrate pathways, both catabolic and anabolic pathways, including fructose, galactose and glycogen metabolism, resulting in a heterogeneous group of disorders. However, data on these pathologies in Colombia are scarce. The aim of our study was to identify genomic variants associated with carbohydrate IEM in the southwestern Colombian population lacking clinical diagnosis. Results: We performed a cross-sectional study, sequencing 320 exomes and classifying variants according to standard guidelines. We identified 286 variants, including 206 new variants and 73 benign or probably benign variants. Notably, six variants in genes such as GALT, GAK1, ALDOB, GAA and SLC2A1 were of uncertain significance, and one pathogenic variant in the GALT gene was associated with classic galactosemia. Interestingly, 18% were intronic, 5% missense, 10% synonymous, and 67% unreported variants.
Conclusions: These findings highlight the need for early diagnostic programs to implement targeted treatments, including transdisciplinary management to minimize morbidity and mortality. Genetic counseling and risk education are crucial, facilitating anticipatory and preventive medicine approaches, thus advancing precision medicine.
INTRODUCTION
Inborn Errors of Metabolism (IEM) of carbohydrates, also known as Hereditary Metabolic Disorders according to the International Classification of Inborn Metabolic Disorders (ICIMD), represent a heterogeneous group of genetic disorders that affect the body's ability to properly process and use carbohydrates, such as disorders of galactose and fructose metabolism, glycogen metabolism and glycolysis; pentose metabolism, transmembrane hexose transport, and absorption [1,2]. Carbohydrate IEMs result from the alteration of catabolic or anabolic pathways of different carbohydrates, with the most common being glucose, fructose, galactose, and glycogen, with prognoses for affected patients ranging from benign to potentially life-threatening [3]. Additionally, existing literature has documented a number of carbohydrate IEMs of great interest, such as galactosemia, fructose intolerance, and glucose-6-phosphate dehydrogenase deficiency (G6PDH), among others [4].
These disorders are the result of variants in key genes involved in carbohydrate metabolic pathways, affecting the synthesis or degradation of these compounds, with differentiated genetic variability according to the population, including the southwest of Colombia, which can lead to specific profiles of genomic variants associated with these congenital errors [5]. These pathologies, although rare at an individual level, are collectively common, with an estimated worldwide incidence of 1 in 1,900 births [6].
The age of onset can vary from childhood to adolescence and even adulthood, although more severe forms tend to manifest during the neonatal period and are associated with significant mortality [7]. In Colombia, the laboratory diagnosis of these metabolic disorders is not a common practice in health services; however, thanks to the joint efforts of health service providers and healthcare professionals, in recent years, significant efforts have been made to intensify the dissemination and knowledge of these disorders [8].
Similarly, in Colombia, neonatal screening programs are being created under Law 1980 of 2019. This law only considers six pathologies, such as phenylketonuria, galactosemia, cystic fibrosis, congenital adrenal hyperplasia, biotinidase deficiency, and hemoglobin disorders, leaving out other IMDs related to carbohydrates, such as Pompe disease (PD). Furthermore, the implementation is moving slowly, as current national screening programs only actively seek congenital hypothyroidism as part of the federally funded programs.
The sequencing of the whole exome (WES) is becoming more common in clinical practice, as it addresses a larger number of genes and is able to identify point mutations and other types of genetic alterations in DNA. This technique has proven its usefulness in the diagnosis of IMDs, and its decreasing cost is turning it into a standard procedure in the diagnostic protocols for these disorders [12]. Studies to identify new genetic variants are expanding the field of knowledge about IMDs, which promotes early detection and timely treatment [13]. However, clinical approaches for diagnosing IMDs are challenging, especially for the rarer variants of the disease, presenting a challenge for clinicians who must be familiar with this group of conditions and learn to identify the typical manifestations of different IMDs [14]. Additionally, Colombia lacks studies that characterize and determine the clinical significance of variants found in population genetic studies, furthermore, there is no
Listen to previous allelic frequency studies that allow estimating the risk of occurrence of these diseases in southwestern Colombia, considering other risk factors such as endogamy associated with geographically isolated populations, which increases the risk of occurrence of these mostly autosomal recessive inherited pathologies [15,16].
In the specific context of the southwest region of Colombia, the prevalence and diversity of genomic variants associated with these congenital errors are areas of study that have been little explored. Therefore, the present study sought to identify, determine, and characterize the genomic variants of genes associated with Inborn Errors of Metabolism (EIM) related to carbohydrates, through the analysis of the results of whole exome sequencing of 320 patients from the southwest region of Colombia found in the genomic database of the Institute of Genomics of Medical Genetics in the city of Cali, who did not have a clinical and/or paraclinical diagnosis of these pathologies, in order to determine their allelic frequencies. This will allow for the expansion of knowledge about these pathologies among the medical and scientific community, as well as health authorities, in order to suggest the establishment of screening and early diagnosis programs, which will allow for the implementation of timely treatments, since there are specific and targeted options available for some of these pathologies.
MATERIALS AND METHODS
This is a cross-sectional, observational, and descriptive study in which the candidate genetic variants of galactose-1-phosphate uridyltransferase (GALT), galactokinase 1 (GALK1), fructose-bisphosphate aldolase B (ALDOB), alpha-glucosidase (GAA), glucose transporter type 1 (SLC2A1), and glucose-6-phosphate dehydrogenase (G6PDH) were sought, identified, and characterized genomically and bioinformatically. These genetic variants were previously identified through literature review, taking into account the frequency of reports, clinical impact, prevalence, and incidence; associated with galactosemia type I and II, hereditary fructose intolerance, Pompe disease, deficiency of glucose transporter I, deficiency of glucose 6-phosphate dehydrogenase in a population in the southwest of Colombia with different pathologies and clinically and/or paraclinically undiagnosed with IEM by carbohydrates.
Literature review
For the selection of genes associated with IMDs (Inborn Errors of Carbohydrate Metabolism), a literature review was conducted in the Scopus, PubMed and LILACS databases for articles published between 2010 and February 2023 (in English, Spanish and Portuguese), including articles specifically related to inborn errors of carbohydrate metabolism, providing information on prevalence, clinical characteristics, and/or information on lethality, and conducted in humans. Two researchers conducted the search independently and a third researcher verified the information. Subsequently, relevant information was recorded in an Excel spreadsheet to organize publications and summarize the main data.
Study population
The results obtained from the whole exome sequencing of 320 patients who did not present clinical and/or paraclinical diagnosis of carbohydrate metabolism disorders from the southwestern region of Colombia were used. These results were provided by the Institute of Medical Genetics - GENOMICS (Cali), with prior signed consent from the participants.
Selection criteria
- Results of whole exome sequencing of patients who did not present clinical and/or paraclinical diagnosis of carbohydrate metabolism disorders from the database of the Institute of Medical Genetics - GENOMICS (Cali).
- Patients from the southwestern regions of Colombia.
- All patients who signed a consent form.
- Informed assent for the use of data.
Exome sequencing
Whole exome sequencing was performed on the patients by extracting blood and collecting it on filter paper and immersing it in phosphate buffer. DNA extraction was carried out using the Qiagen DNeasy kit; the quality and quantity of DNA from each sample were quantified and verified. Next, the Nextera TM libraries were sequenced using the Illumina platform with a coverage of 100X. Sequence alignment was performed with the GRCh38/hg19 reference genome. The sequencing results are in variant call format (VCF) files and underwent a preliminary interpretation and prioritization filter, all with the aim of ensuring the accuracy and quality of the genomic data obtained.
Variant identification
All exomes belonging to the big data of the Institute of Medical Genetics - GENOMICS (Cali), a total of 320 patients, are in VCF (Variant Call Format) format. For the identification of genetic variants, the VCF Editor 1.0 program was used, which allows processing the VCF format of the exomes, applying different search filters, and obtaining a final spreadsheet file with information organized in columns by chromosome, position, altered nucleotide, Clinvar record, among other characteristics [17].
Bioinformatic analysis
Bioinformatic analysis of the variants was performed using the Clinvar, Varsome, and Intervar databases to identify if they had been previously described [18-20]. Furthermore, the disease-associated consequences were determined using bioinformatic tools, Functional Analysis through Hidden Markov Models (FATHMM), Polymorphism Phenotyping v2 (Polyphen-2), Mutation Taster, MutationAssessor, Protein Variation Effect Analyzer (PROVEAN), Sorting Intolerant From Tolerant (SIFT), UMD-Predictor, Human Splicing Finder.
Classification of variants
Genetic variants were classified according to the guidelines for the interpretation of sequence variants from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [21].
Calculation of allele frequencies
Allelic frequency was calculated by simple counting, dividing the number of total alleles found for each variant by the total number of patients with exome.
Equation 1. Allelic frequency by simple count [22]:
Number of copies of the allele in the population / Total number of copies of the gene in the population
Ethical aspects
The ethics committee of the Universidad del Valle (Cali, Colombia) reviewed and approved the study. The authors declare no conflicts of interest. Furthermore, in this article, the confidentiality of the data was maintained, and informed consent was obtained from the legal representative of the patient (father and mother).
RESULTS
Literature review
The search strategy yielded 706 records, after excluding duplicate records and applying inclusion criteria, 94 publications were obtained, 47 were excluded after full-text analysis, and 47 studies were included in the final analysis, which most frequently reported data on the genes GALT, GALK1, ALDOB, GAA, SLC2A1, and G6PD, associated with IMDs by carbohydrates; therefore, these genes were chosen to be evaluated in the study population.
Genomic Variants in Carbohydrate IMDs
Through genomic and bioinformatic analysis of 320 complete exomes from patients in southwestern Colombia, without clinical and/or paraclinical diagnosis associated with inborn errors of carbohydrate metabolism, 286 variants related to the genes ALDOB, G6PD, GAA, GALK1, GALT, and SLC2A1 were found; 191 of these variants had not been previously reported in the Clinvar, Intervar, and Varsome databases, 19 of them do not have reported clinical significance in these platforms. The genes with the highest number of variants found were GALK1, GAA, and G6PD associated with galactosemia type II, Pompe disease, and glucose-6-phosphate dehydrogenase deficiency respectively; the genes with the lowest number of reported variants were ALDOB and GALT. Among the variants that had been previously reported, 29% were synonymous changes, 16% were missense changes, and 55% were variants in non-coding regions in the genes associated with carbohydrate IMDs (Figure 1).
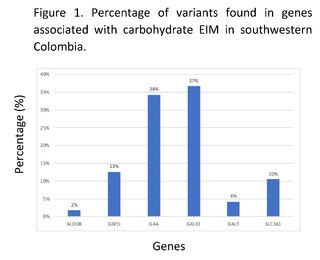
Clinical significance of genomic variants in genes associated with IBD by carbohydrates in the Colombian southwest.
Regarding the clinical significance given by the ACMG and according to the prediction software used in the variants found, 24.5% presented a benign and probably benign meaning, 3.1% presented an uncertain meaning, and 0.3% were classified as pathogenic or probably pathogenic. For this last classification, in the GAA gene, the modeling and prediction software showed a pathogenic effect in three variants (p.Gly576Ser, p.Asp91Asn, and p.Thr927Ile); however, due to their very high allelic frequency, 2.5%, 2.2%, and 1.6% respectively were reclassified as benign and probably benign. The remaining 72% corresponded to variants that have not been functionally reported and their clinical importance is currently unknown.

Allelic frequencies found
According to the allelic frequencies analyzed, the variant at position chr17:g.73753661C>T in the GALK1 gene had the highest frequency, with 34.38% in the entire sample; however, this variant did not show any reported clinical significance in databases such as Clinvar and Varsome. Similarly, the variant c.*22+1116G>C in the GALK1 gene had a frequency of 32.19%, which has not been reported in databases like Clinvar, nor in intronic region prediction programs like Human Splincing Finder, however, it did show a benign clinical significance in other databases such as Varsome.
The variants that were analyzed by the prediction software and classified as pathogenic, probably pathogenic, or pseudodeficient variants, as shown in Table 2, had an allelic frequency ranging from 0.025 to 0.00625, with 0.00625 being the lowest frequency reported for the p.Ser135Leu variant in the GALT gene.

DISCUSSION
In the complete exomes of 320 patients with complex and undiagnosed clinical and/or paraclinical pathologies of any IEM, 286 variants were found for the genes chosen by the literature: GALT, GALK1, ALDOB, GAA, G6PD, and SLC2A1, of which, according to the criteria established in the guidelines issued by the ACMG and the Association for Molecular Pathology, were classified into 73 variants of benign or likely benign significance, 1 variant of pathogenic significance, 6 variants of uncertain significance, and 206 variants that have not been previously reported in clinical and population databases.
Pathogenic variant in GALT
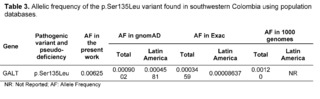
Classical Galactosemia (CG) belongs to a rare group of hereditary disorders of galactose metabolism, caused by defects in the enzyme galactose-1-phosphate uridyltransferase encoded by the GALT gene; as a consequence of the enzymatic deficit, patients present a wide spectrum of physical and cognitive disabilities, potentially leading to mortality [23]. Regarding the GALT gene, within the results of the 320 complete exomes, the variant c.404C>T (p.Ser135Leu) had a clinical significance of pathogenicity through the analysis performed by the bioinformatics software, with a frequency of 0.00625, which has been described to mainly affect patients of African descent and comprises up to 90% of the causal variants of galactosemia in South African patients [24,25]. The p.Ser135Leu variant demonstrated a pathogenic effect after in silico protein functional analysis in all the prediction software programs used, which is consistent with the in silico studies conducted by Kumar S U et al., that had also confirmed the pathogenic effect of this variant.
They used a comprehensive computational process for the nonsense variant, p.Ser135Leu, to analyze the conservation of amino acids, pathogenicity predictions, and protein stability, using bioinformatics tools. They found that the substitution of the native amino acid with a leucine causes the loss of hydrogen bonds in the protein core, altering the correct folding in the loop region and the eight beta-sheet regions. They concluded that p.Ser135Leu, compared to other variants, is the most deleterious among prevalent variants that alter the overall function of the protein [26].
This is also supported by studies conducted by Coelho AI et al., when they molecularly characterized GALT deficiency in Portugal, in forty-two galactosemic patients where the p.Ser135Leu variant was the third most prevalent in their population (4%), the parents of patients carrying the p.Ser135Leu variant were of African origin [27]. Studies conducted by Crespo C, et al., in 2020 in 37 Argentinian patients with a galactose-1-phosphate uridylyltransferase enzymatic activity lower than 35% of the normal value, identified the p.Ser135Leu variant in a patient with zero enzymatic activity [28].
The allelic frequency of the p.Ser135Leu variant was higher than the reported Latin frequencies by ExAC and gnomAD; likewise, this variant in the study population exceeded the data reported in global frequencies (Table 3). The prevalence of African variants in the population of southwestern Colombia may be associated with the high percentage of African ancestry in Colombian territory. It can vary between 0.0 and 92.1% depending on the population and ethnic group of the study [29].
Pseudodeficiency variants in OAG
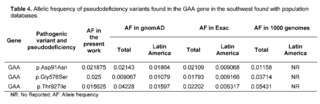
Pathological alterations in the protein product of the GAA gene are associated with type II glycogen storage disease or EP, caused by a decrease in alpha-glucosidase acid, necessary for the degradation of glycogen into glucose, altering the function of cellular and lysosomal metabolism. [30]. Variants of the GAA gene represented 36% of the variants found in the present study, where 50% of the variants were found in intronic regions. Regarding the variants found, the p.Gly576Ser variant of common pseudodeficiency [31] has been described by Reuser AJJ et al. in heterozygosity in a cis position in 17 Asian patients with classical infantile EP with less severe cardiomyopathies and slower disease progression [32]. Furthermore, Reuser AJJ et al. found other pseudodeficiency variants; p.Asp91Asn and p.Thr927Ile in Caucasian patients affected with late-onset EP without cardiomyopathy, with a more attenuated form with symptom onset before the age of 12 [32].
The pseudodeficiency alleles p.Asp91Asn and p.Gly576Ser have been reported in an American population of Asian descent, in 38 infants with positive screening for Pompe disease, with late-onset being more prevalent [33]. Similarly, the variants p.Asp91Asn and p.Gly576Ser were described in 3 patients with suspected late-onset Pompe disease, presenting other altered biochemical tests along with enzymatic activity very close to the minimum values [34] with varying degrees of sarcopenia or frailty syndrome in adulthood [35].
In comparison to population databases like 1,000 Genomes, ExAC, and gnomAD, the variant p.Aps91Asn for GAA was found to have an allelic frequency within global frequency values but well above what is reported for Latin America, GnomAD, and Exac. The variant Gly576Ser for GAA had an allelic frequency above that reported in population databases containing gene frequencies of Latin American populations, and this variant also had a much higher prevalence in the Latino population compared to the global frequency in these databases (Table 4).
In Colombia, Pompe disease is not included in neonatal screening programs, although it can be identified through tandem mass spectrometry [33]. In addition to this, the benefits of treatment before the onset of clinical symptoms in this progressive disease have been documented, avoiding irreversible muscle damage [36]. The results of variability and allelic frequency of the GAA gene are the basis for alerting the competent health authorities about the existence of these variants in the population of southwestern Colombia, recognizing the importance of including this pathology in neonatal screening programs.
Variants of pseudodeficiency in G6PDH
G6PDH deficiency is considered the most common genetic abnormality that is inherited as a polymorphic trait linked to the X chromosome, with an estimated 400 million affected individuals and its distribution and genetic variability is closely related to geographical regions and ethnic groups [37]. In relation to the present study, the genetic variability of G6PD was low in the 320 complete exomes of patients without clinical and/or paraclinical diagnosis belonging to the southwest region of Colombia, with a total of 36 variants, of which 29 variants have not been previously reported, 4 variants had a clinical significance of benign/probably benign, and 3 variants had an uncertain clinical significance.
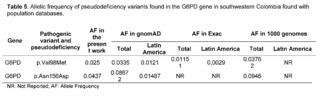
The variant of uncertain significance c.292G>A, p.Val98Met, also known as the G6PD A- variant (rs1050828), has been described in African populations as producing 10 to 60% of normal enzymatic activity. It had an allelic frequency of 0.025 in the population of southwestern Colombia. However, the homozygous variant p.Val98Met is not sufficient to cause enzymatic deficiency and requires the association with the variant c.466A>G, p.Asn156Asp. This variant had a frequency of 0.043 in the present study, being a higher frequency than that reported for Latin America in databases such as gnomAD and 1000 Genomes (Table 5).
Valencia SH et al. studies also assessed the prevalence of G6PD deficiency in 426 individuals through enzymatic activity analysis and genotyping in endemic malaria regions of the Colombian Pacific coast [40]. In these regions located in the southwestern part of Colombia, Valencia SH et al. reported the existence of the G6PDD A- variants; p.Val98Met and p.Asn156Asp with severe to intermediate enzymatic deficiency in 6.62% of hemizygous men and 4.64% in women; and along with the A+ variant which only includes; c.376 A>G, reported an intermediate enzymatic deficiency in five hemizygous men and one heterozygous woman, with the highest proportion of individuals with haplotypes A- and A+ of G6PD, coming from Tumaco and Buenaventura, regions related to the study population. Vela-Amieva Met al. studies, in a cohort of 81 Mexican newborns, in a neonatal screening program for G6PDd found nine different G6PDd genotypes; with G6PD A- p.Val98Met and p.Asn156Asp being the most frequent (60.5%), demonstrating that
The same variant of G6PD could cause a very variable residual enzymatic activity, changing the course of the phenotypic spectrum even from the first day of life, as the average residual G6PD activity percentage in hospitalized patients in the neonatal period with or without neonatal jaundice varied between 29 and 34% [42].
In-silico assays conducted by Batista da Rocha J. et al. evaluated the functional impact of the African variants, p.Val98Met and p.Asn156Asp, using structural bioinformatics techniques. They have described significant changes in the residues of the catalytic binding domain of NADP+, in terms of direction and impact, highlighting that the differences are of an additive nature, in relation to the NADP+ and G6PD binding, finding greater changes with the p.Val98Met variant in contrast to the p.Asn156Asp variant. However, in the presence of both variants, the G6PD A- genotype showed greater instability in enzymatic function compared to the p.Val98Met variant alone [43].
CONCLUSION
In the studied population, a high frequency of the variants p.Val98Met and p.Asn156Asp associated with G6PD deficiency was found, which has an uncertain clinical significance in the literature and its presence in carriers is associated with G6PD deficiency. These findings suggest that these variants in the G6PD gene could be considered a risk factor for the population of southwestern Colombia. Additionally, with a lower allelic frequency, the pathogenic variant p.Ser135Leu in the GALT gene associated with classical galactosemia circulates within southwestern Colombia, suggesting that conditions such as galactosemia may contribute to the diagnostic burden of IMDs related to carbohydrate metabolism within the Colombian territory.
This work increased the information reported so far on the variants associated with IMDs by carbohydrates, allowing for future research, recognizing the impact of pathogenic variants and pseudodeficiency that can occur in society. It also contributed to the knowledge and creation of molecular bases for IMDs by carbohydrates in southwestern Colombia, allowing for adequate genetic counseling and education on the risk of heritability, practicing anticipatory and preventive medicine, getting closer to precision medicine.
The use of databases and bioinformatics software allowed us to predict the clinical significance of the different variants, from genotype to phenotype. This research not only provides a deeper understanding of genetic variability in the studied population, but also aims to be a valuable reference for similar research in different geographical contexts. This comprehensive approach can contribute to the development of public health and personalized strategies in the identification and management of carbohydrate IMDs worldwide.
ACKNOWLEDGEMENTS
Thanks to the participating patients, the Institute of Medical Genetics Genomics (GENOMICS), the University of Valle, and the Research Group on Congenital Metabolic Diseases.
AUTHOR CONTRIBUTIONS
JDVB: Research, LJMG: Supervision, JMSS: Supervision.
References
[1] Stenton SL, Kremer LS, Kopajtich R, Ludwig C, Prokisch H. The diagnosis of inborn errors of metabolism by an integrative “multi‐omics” approach: A perspective encompassing genomics, transcriptomics, and proteomics. J Inherit Metab Dis [Internet]. 2020 Jan 25;43(1):25–35. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jimd.12130
[2] Ferreira CR, Rahman S, Keller M, Zschocke J. An international classification of inherited metabolic disorders ( ICIMD ). J Inherit Metab Dis [Internet]. 2021 Jan 27;44(1):164–77. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jimd.12348
[3] Ferguson AM. Chapter 7 - Carbohydrate disorders. In: Garg U, Smith LDBT-B in IE of M, editors. San Diego: Elsevier; 2017. p. 155–66. Available from: http://www.sciencedirect.com/science/article/pii/B9780128028964000134
[4] Ferreira CR, van Karnebeek CDM, Vockley J, Blau N. A proposed nosology of inborn errors of metabolism. Genet Med [Internet]. 2019 Jan 8;21(1):102–6. Available from: http://www.nature.com/articles/s41436-018-0022-8
[5] Guo K, Zhou X, Chen X, Wu Y, Liu C, Kong Q. Expanded Newborn Screening for Inborn Errors of Metabolism and Genetic Characteristics in a Chinese Population. Front Genet [Internet]. 2018 Apr 20;9. Available from: http://journal.frontiersin.org/article/10.3389/fgene.2018.00122/full
[6] Tebani A, Abily-Donval L, Afonso C, Marret S, Bekri S. Clinical Metabolomics: The New Metabolic Window for Inborn Errors of Metabolism Investigations in the Post-Genomic Era. Int J Mol Sci [Internet]. 2016 Jul 20;17(7):1167. Available from: http://www.mdpi.com/1422-0067/17/7/1167
[7] Moreno-Giraldo LJ, Satizábal-Soto JM. Aproximación Diagnóstica de los Errores Innatos del Metabolismo. In: “Estructura del genoma humano y las enfermedades genéticas” Semiología pediatrica. Colombia: Editorial Distribuna; 2021. p. 511–24.
[8] Puentes-Tellez MA, Lerma-Barbosa PA, Garzón-Jaramillo RG, Suarez DA, Espejo-Mojica AJ, Guevara JM, et al. A perspective on research, diagnosis, and management of lysosomal storage disorders in Colombia. Heliyon [Internet]. 2020 Mar;6(3):e03635. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2405844020304801
[9] Congreso de la República de Colombia. Ley 1990 De 2019. 2019;(51):17–2012.
[10] Ministerio de Salud y Protección Social. Resolución 5265 De 2018 Listado de Enfermedades Huérfanas. Minist Salud y Protección Soc [Internet]. 2018;26. Available from: http://achc.org.co/wp-content/uploads/2018/12/RESOLUCIÓN-No.-5265-DE-2018-MINSALUD-LISTADO-ENFERMEDADES-HUÉRFANAS..pdf
[11] Avellaneda LAB. Los Errores Innatos del Metabolismo, fuente inagotable de conocimiento médico por más de un siglo, siguen sin diagnosticarse y tratarse en Colombia. NOVA. 2005;3(3).
[12] Shakiba M, Keramatipour M. Effect of Whole Exome Sequencing in Diagnosis of Inborn Errors of Metabolism and Neurogenetic Disorders. Iran J child Neurol. 2018;12(1):7–15.
[13] González-Lamuño Leguina D, Bóveda Fontán MD, Bueno Delgado M, Gort Mas L, Unceta Suárez M, Morales Conejo M. [The metabolic newborn screening as a healthcare model of the precision medicine. Perspective from the Spanish Association for the Study of Congenital Errors of Metabolism (AECOM).]. Rev Esp Salud Publica [Internet]. 2021 Jan 26;95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33496277
[14] Ferreira CR, van Karnebeek CDM. Inborn errors of metabolism. In 2019. p. 449–81. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780444640291000229
[15] Afzal RM, Lund AM, Skovby F. The impact of consanguinity on the frequency of inborn errors of metabolism. Dan Med J [Internet]. 2018 Oct;65(10). Available from: http://www.ncbi.nlm.nih.gov/pubmed/30269750
[16] Guauque-olarte S, Sc B, Patricia Á. DIVERSIDAD Y ESTRUCTURA GENÉTICA DE TRES POBLACIONES AFRODESCENDIENTES DEL SUROCCIDENTE COLOMBIANO A PARTIR DE 8 STR’S. 2010;15:47–60.
[17] Moreno-Giraldo LJ, Satizábal-Soto JM, Sanchéz-Gomez A. Research Article Genomic variability of the mucopolysaccharidosis complex in southwestern Colombia. Genet Mol Res [Internet]. 2020;19(2). Available from: http://www.funpecrp.com.br/gmr/articles/year2020/vol19-2/pdf/gmr18502_-_genomic-variability-mucopolysaccharidosis-complex-southwestern-colombia.pdf
[18] Landrum MJ, Kattman BL. ClinVar at five years: Delivering on the promise. Hum Mutat [Internet]. 2018 Nov 11;39(11):1623–30. Available from: https://onlinelibrary.wiley.com/doi/10.1002/humu.23641
[19] Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019 Jun;35(11):1978–80.
[20] Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet [Internet]. 2017 Feb;100(2):267–80. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002929717300046
[21] Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med [Internet]. 2015 May 5;17(5):405–23. Available from: http://www.nature.com/articles/gim201530
[22] Andrews C. The Hardy-Weinberg Principle. 2010;10(Nature Education Knowledge):65.
[23] Banford S, McCorvie TJ, Pey AL, Timson DJ. Galactosemia: Towards Pharmacological Chaperones. J Pers Med [Internet]. 2021 Feb 7;11(2):106. Available from: https://www.mdpi.com/2075-4426/11/2/106
[24] Henderson H, Leisegang F, Brown R, Eley B. The clinical and molecular spectrum of galactosemia in patients from the Cape Town region of South Africa. BMC Pediatr [Internet]. 2002 Dec 2;2(1):7. Available from: http://bmcpediatr.biomedcentral.com/articles/10.1186/1471-2431-2-7
[25] Suzuki M, West C, Beutler E. Large-scale molecular screening for galactosemia alleles in a pan-ethnic population. Hum Genet [Internet]. 2001 Aug 1;109(2):210–5. Available from: http://link.springer.com/10.1007/s004390100552
[26] Kumar S U, Kumar D T, R S, Doss C GP, Zayed H. An extensive computational approach to analyze and characterize the functional mutations in the galactose-1-phosphate uridyl transferase (GALT) protein responsible for classical galactosemia. Comput Biol Med [Internet]. 2020 Feb;117:103583. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0010482519304366
[27] Coelho AI, Ramos R, Gaspar A, Costa C, Oliveira A, Diogo L, et al. A frequent splicing mutation and novel missense mutations color the updated mutational spectrum of classic galactosemia in Portugal. J Inherit Metab Dis [Internet]. 2014 Jan 8;37(1):43–52. Available from: http://doi.wiley.com/10.1007/s10545-013-9623-1
[28] Crespo C, Eiroa H, Otegui MI, Bonetto MC, Chertkoff L, Gravina LP. Molecular analysis of GALT gene in Argentinian population: Correlation with enzyme activity and characterization of a novel Duarte-like allele. Mol Genet Metab Reports [Internet]. 2020 Dec;25:100695. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2214426920301415
[29] Chande AT, Nagar SD, Rishishwar L, Mariño-Ramírez L, Medina-Rivas MA, Valderrama-Aguirre AE, et al. The Impact of Ethnicity and Genetic Ancestry on Disease Prevalence and Risk in Colombia. Front Genet [Internet]. 2021 Sep 17;12. Available from: https://www.frontiersin.org/articles/10.3389/fgene.2021.690366/full
[30] Costa-Verdera H, Collaud F, Riling CR, Sellier P, Nordin JML, Preston GM, et al. Hepatic expression of GAA results in enhanced enzyme bioavailability in mice and non-human primates. Nat Commun [Internet]. 2021 Dec 4;12(1):6393. Available from: https://www.nature.com/articles/s41467-021-26744-4
[31] Yang C-C, Chien Y-H, Lee N-C, Chiang S-C, Lin S-P, Kuo Y-T, et al. Rapid progressive course of later-onset Pompe disease in Chinese patients. Mol Genet Metab [Internet]. 2011 Nov;104(3):284–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1096719211001971
[32] Reuser AJJ, Ploeg AT, Chien Y, Llerena J, Abbott M, Clemens PR, et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum Mutat [Internet]. 2019 Nov 7;40(11):2146–64. Available from: https://onlinelibrary.wiley.com/doi/10.1002/humu.23878
[33] Burton BK, Charrow J, Hoganson GE, Fleischer J, Grange DK, Braddock SR, et al. Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. Int J Neonatal Screen [Internet]. 2020 Jan 21;6(1):4. Available from: https://www.mdpi.com/2409-515X/6/1/4
[34] Ficicioglu C, Ahrens-Nicklas RC, Barch J, Cuddapah SR, DiBoscio BS, DiPerna JC, et al. Newborn Screening for Pompe Disease: Pennsylvania Experience. Int J Neonatal Screen [Internet]. 2020 Nov 13;6(4):89. Available from: https://www.mdpi.com/2409-515X/6/4/89
[35] Palmio J, Udd B. Borderlines between Sarcopenia and Mild Late-Onset Muscle Disease. Front Aging Neurosci [Internet]. 2014 Sep 29;6. Available from: http://journal.frontiersin.org/article/10.3389/fnagi.2014.00267/abstract
[36] de Las Heras J, Cano A, Vinuesa A, Montes M, Unceta Suarez M, Arza A, et al. Importance of Timely Treatment Initiation in Infantile-Onset Pompe Disease, a Single-Centre Experience. Child (Basel, Switzerland). 2021 Nov;8(11).
[37] Sathupak S, Leecharoenkiat K, Kampuansai J. Prevalence and molecular characterization of glucose-6-phosphate dehydrogenase deficiency in the Lue ethnic group of northern Thailand. Sci Rep [Internet]. 2021 Dec 3;11(1):2956. Available from: http://www.nature.com/articles/s41598-021-82477-w
[38] Walakira A, Tukwasibwe S, Kiggundu M, Verra F, Kakeeto P, Ruhamyankaka E, et al. Marked variation in prevalence of malaria-protective human genetic polymorphisms across Uganda. Infect Genet Evol [Internet]. 2017 Nov;55:281–7. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1567134817303271
[39] Ouattara AK, Yameogo P, Traore L, Diarra B, Assih M, Compaore TR, et al. Prevalence, genetic variants and clinical implications of G-6-PD deficiency in Burkina Faso: a systematic review. BMC Med Genet [Internet]. 2017 Dec 23;18(1):139. Available from: https://bmcmedgenet.biomedcentral.com/articles/10.1186/s12881-017-0496-2
[40] Valencia SH, Ocampo ID, Arce-Plata MI, Recht J, Arévalo-Herrera M. Glucose-6-phosphate dehydrogenase deficiency prevalence and genetic variants in malaria endemic areas of Colombia. Malar J [Internet]. 2016 Dec 26;15(1):291. Available from: https://malariajournal.biomedcentral.com/articles/10.1186/s12936-016-1343-1
[41] Carmona-fonseca J, Alvarez G, Rios A, Vasquez MF. Deshidrogenasa en hombres sanos y en pacientes maláricos ; Turbo ( Antioquia , Colombia ) dehydrogenase in healthy men and. Rev Bras Epidemiol. 2008;11(2):252–65.
[42] Vela-Amieva M, Alcántara-Ortigoza MA, González-del Angel A, Belmont-Martínez L, López-Candiani C, Ibarra-González I. Genetic spectrum and clinical early natural history of glucose-6-phosphate dehydrogenase deficiency in Mexican children detected through newborn screening. Orphanet J Rare Dis [Internet]. 2021 Dec 26;16(1):103. Available from: https://ojrd.biomedcentral.com/articles/10.1186/s13023-021-01693-9
[43] Batista da Rocha J, Othman H, Hazelhurst S. Molecular dynamics of G6PD variants from sub-Saharan Africa. Biochem Biophys Reports [Internet]. 2022 Jul;30:101236. Available from: https://linkinghub.elsevier.com/retrieve/pii/S240558082200036
