
Cuando se duplica el cromosoma 15: un reto clínico-genético
 ,
,
Autores/as
DOI:
https://doi.org/10.37980/im.journal.ggcl.es.20252717Palabras clave:
Síndrome dup15q, cromosoma 15q11-q13, array-CGH, autism , spectrum disorder, epilepsyResumen
Antecedentes: El síndrome de duplicación del cromosoma 15q11–q13 (Dup15q) es un trastorno neurodel desarrollo poco frecuente causado por la presencia de copias adicionales de una región genómica enriquecida en genes imprintados esenciales para la función neuronal normal. Se asocia con frecuencia con trastorno del espectro autista (TEA), epilepsia, hipotonía, retraso global del desarrollo y discapacidad intelectual. Presentación del caso: Se reporta el caso de un varón de 31 años con antecedentes de epilepsia refractaria, trastorno del espectro autista y discapacidad intelectual. La evaluación genética mediante hibridación genómica comparativa por microarreglos (array-CGH), combinada con análisis de polimorfismos de un solo nucleótido (SNP), identificó una duplicación intersticial heterocigota de novo de 10,03 Mb en la región 15q11.2–q13.3. El segmento duplicado incluye múltiples genes con funciones clave en el neurodesarrollo, entre ellos UBE3A, GABRB3, GABRA5, SNRPN y NDN. El estudio parental mediante amplificación dependiente de ligación de sondas múltiples (MLPA) fue negativo, lo que confirmó un origen no heredado. El establecimiento del diagnóstico molecular permitió una clasificación clínica precisa, orientó el manejo terapéutico y facilitó el asesoramiento genético. Conclusión: Este caso ilustra la utilidad diagnóstica del array-CGH en pacientes con fenotipos neurodel desarrollo complejos y subraya la importancia de la evaluación genética temprana en individuos con TEA y epilepsia. La identificación de variantes patogénicas en el número de copias, como el Dup15q, tiene implicaciones directas en el pronóstico, la planificación terapéutica y el asesoramiento familiar.
Introducción
El síndrome de duplicación 15q11.2–q13.3 (Dup15q) es un trastorno neurodel desarrollo poco frecuente, de herencia autosómica dominante, causado por la duplicación del segmento cromosómico 15q11.2–q13.3. Esta región, localizada en el brazo largo del cromosoma 15, es altamente susceptible a errores de recombinación durante la meiosis debido a la presencia de secuencias repetidas de bajo número de copias (LCRs). Estas anomalías estructurales surgen principalmente a través de recombinación homóloga no alélica (NAHR) entre regiones específicas de puntos de ruptura (BP1 a BP5) [1].
La duplicación puede presentarse en dos formas principales: como una duplicación intersticial en el cromosoma 15 de origen materno o como un cromosoma isodicéntrico supernumerario [idic(15)], que implica dos copias adicionales de la región duplicada. Estas variantes confieren una expresividad clínica variable y penetrancia incompleta; algunos portadores pueden ser asintomáticos o presentar un fenotipo leve. Las duplicaciones de origen materno se han asociado con manifestaciones clínicas más severas en comparación con las de origen paterno [1].
Desde una perspectiva fisiopatológica, se ha propuesto que el desequilibrio en la dosis génica causado por la duplicación contribuye a la sobreexpresión patológica de genes críticos para el desarrollo cerebral. Este fenómeno puede interferir con la maduración sináptica, la plasticidad neuronal y la regulación de los canales iónicos, procesos esenciales para el funcionamiento adecuado del sistema nervioso central. Estas alteraciones neurobiológicas subyacen a los signos y síntomas característicos del síndrome [2].
Clínicamente, el Dup15q presenta un amplio espectro de manifestaciones que incluyen retraso global del desarrollo, discapacidad intelectual, hipotonía neonatal, alteraciones del lenguaje y de las habilidades motoras, trastorno del espectro autista (TEA) y epilepsia farmacorresistente, esta última asociada a un mayor riesgo de muerte súbita inesperada en epilepsia (SUDEP). La epilepsia es una de las manifestaciones más incapacitantes, afecta a más del 60 % de los pacientes y suele presentarse con crisis polimorfas que pueden evolucionar hacia un fenotipo tipo Lennox–Gastaut. Los hallazgos electroencefalográficos pueden incluir puntas focales, puntas generalizadas, ritmos rápidos difusos e hipsarritmia. También pueden observarse rasgos dismórficos leves como nariz respingada, pliegues epicánticos y fisuras palpebrales oblicuas hacia abajo [1–5].
El diagnóstico comienza con un alto índice de sospecha clínica tras el reconocimiento de estos signos. Debido a la superposición fenotípica con otros trastornos genéticos como los síndromes de Angelman o Prader–Willi, se requieren herramientas genéticas de alta resolución para confirmar el diagnóstico. El microarreglo cromosómico o la hibridación genómica comparativa (CMA o array-CGH) se consideran pruebas de primera línea, ya que permiten la detección de duplicaciones submicroscópicas y proporcionan una localización y delimitación precisas de la anomalía. Esta recomendación ha sido respaldada por múltiples guías internacionales, incluidas las del American College of Medical Genetics and Genomics [6–10].
Una vez detectada la duplicación, se recomiendan técnicas complementarias para determinar el mecanismo subyacente. La hibridación in situ fluorescente (FISH) y los estudios citogenéticos convencionales permiten detectar la presencia de cromosomas idic(15) e identificar posible mosaicismo. Además, la MLPA (Multiplex Ligation-dependent Probe Amplification) es útil para cuantificar duplicaciones o triplicaciones en la región 15q11.2–q13.3. Finalmente, el análisis de metilación del ADN es esencial para determinar el origen parental de la duplicación, ya que las de origen materno se asocian con un fenotipo clínico más severo [6,8,11,12].
En cuanto al tratamiento, la detección temprana de la duplicación permite la implementación de intervenciones personalizadas dirigidas a optimizar el desarrollo neurocognitivo y mejorar el manejo de comorbilidades como la epilepsia refractaria. Además, los modelos celulares derivados de pacientes, como las células madre pluripotentes inducidas (iPSC), han abierto nuevas vías para investigar los mecanismos moleculares del Dup15q, con el objetivo de desarrollar terapias dirigidas adaptadas a las necesidades específicas de esta población [13].
Presentación del caso
El paciente es un varón de 31 años evaluado por el servicio de genética. Nació de una madre de 34 años y un padre de 35 años al momento de la concepción. La madre tenía antecedente de un aborto espontáneo previo. El embarazo cursó sin exposición conocida a agentes teratogénicos. El parto fue por cesárea a las 42 semanas de gestación debido a embarazo postérmino y se complicó con circular de cordón, líquido amniótico meconial y signos clínicos de asfixia perinatal. El paciente requirió intubación orotraqueal al nacer, aunque no se evidenció aspiración meconial.
No se reportó consanguinidad parental. Los antecedentes familiares revelaron dos tíos paternos con epilepsia no especificada y un tío materno con un trastorno conductual no especificado. No se documentaron antecedentes familiares de malformaciones congénitas, trastornos genéticos, síndromes conductuales ni enfermedades psiquiátricas mayores.
Desde la infancia temprana, el paciente presentó retraso global del desarrollo, con dependencia completa para las actividades básicas de la vida diaria. Las crisis epilépticas comenzaron a los seis años de edad, predominantemente durante el sueño, y se caracterizaron por crisis tónicas generalizadas, sialorrea, relajación de esfínteres y somnolencia posictal. La frecuencia y severidad de las crisis aumentaron progresivamente, con pobre respuesta a múltiples fármacos antiepilépticos, configurando un cuadro de epilepsia refractaria.
El examen físico no evidenció dismorfias faciales evidentes. El paciente deambulaba de forma independiente con marcha estable, sin requerir dispositivos de asistencia. Se observó contacto visual limitado, orejas de implantación anterior, ausencia de lenguaje verbal con emisión de sonidos guturales, aleteo de manos y estereotipias motoras que involucraban extremidades superiores y cabeza. No se identificaron deformidades de la columna ni asimetrías en las extremidades. Los genitales externos eran normales; el prepucio era redundante con un anillo constrictor a nivel del glande y ambos testículos se encontraban en el escroto. No se observó macroorquidismo.
Dada la constelación clínica de epilepsia refractaria, trastorno del espectro autista y discapacidad intelectual, se sospechó fuertemente una etiología genética. Por ello, se realizó un estudio molecular mediante hibridación genómica comparativa basada en microarreglos (array-CGH) integrada con análisis de polimorfismos de un solo nucleótido (SNP). La prueba se realizó utilizando la plataforma SurePrint G3 Human CGH+SNP 4×180K (Agilent Technologies). Esta metodología permite la detección simultánea de variaciones en el número de copias genómicas, como deleciones (pérdidas), duplicaciones (ganancias) y reordenamientos cromosómicos desbalanceados, proporcionando información de alta resolución sobre la arquitectura genómica subyacente a los trastornos del neurodesarrollo.
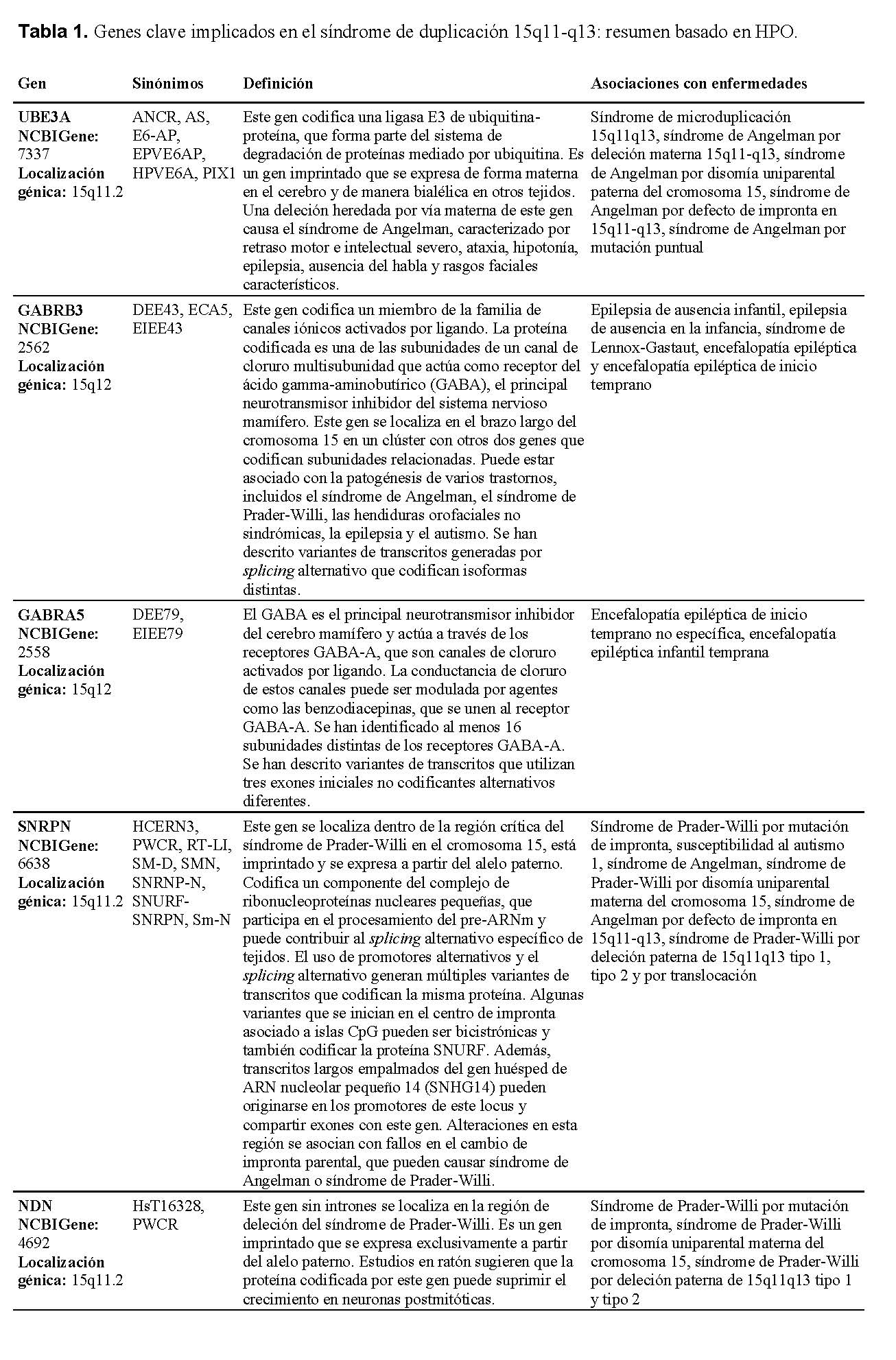
El análisis identificó una duplicación intersticial heterocigota de 10.03 megabases (Mb) en la región 15q11.2–q13.3 (coordenadas genómicas chr15:22572809–32607357, genoma de referencia GRCh38) (Figuras 1 y 2). Este segmento duplicado incluye varios genes funcionalmente relevantes, entre ellos UBE3A, GABRB3, GABRA5, SNRPN y NDN, todos con roles críticos en el neurodesarrollo y la neurotransmisión GABAérgica. El síndrome de duplicación 15q11–q13 (OMIM #608636) se ha asociado con un espectro de condiciones neurodel desarrollo y neuropsiquiátricas, como trastorno del espectro autista, discapacidad intelectual, hipotonía, ataxia, convulsiones, retraso del desarrollo, alteraciones conductuales y esquizofrenia (PMID: 21324950).

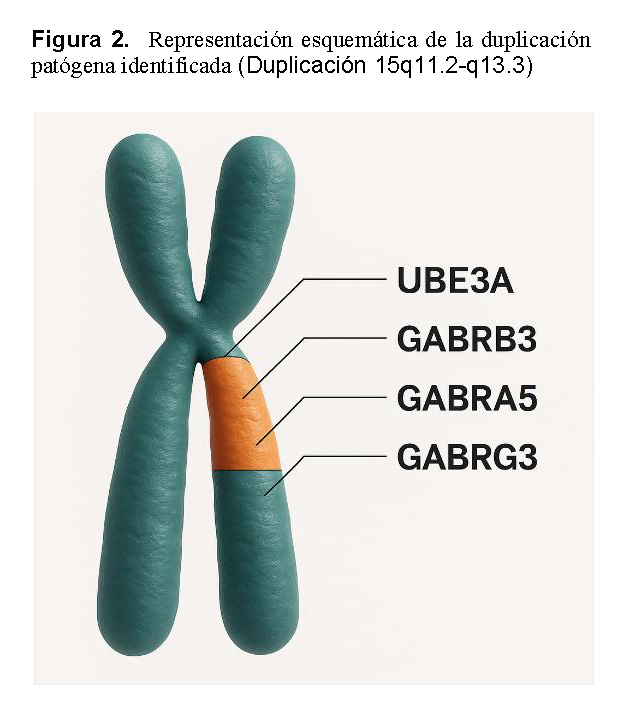
Existe evidencia creciente de un efecto dependiente del origen parental, con duplicaciones de origen materno más frecuentemente asociadas a manifestaciones clínicas. En contraste, las duplicaciones de origen paterno se han observado con mayor frecuencia en individuos no afectados, aunque algunos casos se han descrito en pacientes con trastorno del espectro autista (PMID: 23495136) y convulsiones (PMID: 23633446). Sin embargo, estos hallazgos son inconsistentes y el número actual de casos reportados es insuficiente para establecer de manera concluyente una correlación genotipo–fenotipo en duplicaciones heredadas de origen paterno (PMID: 18840528). De acuerdo con las guías del American College of Medical Genetics and Genomics (ACMG), esta variante en el número de copias se clasifica como patogénica. La tabla 1 resume las características de los genes según la Human Phenotype Ontology.
La identificación de esta duplicación confirmó el diagnóstico de síndrome de duplicación 15q11–q13 (Dup15q). Para investigar el origen genético, se realizó un análisis MLPA dirigido a la región 15q11–q13 en ambos progenitores, con resultados negativos. Este hallazgo respalda un origen de novo de la duplicación y confirma una etiología genética no heredada, basada en la correlación observada entre genotipo, endotipo y fenotipo.
Este diagnóstico permitió una clasificación clínica precisa, orientó la estrategia terapéutica, facilitó una estimación pronóstica más adecuada y sentó las bases para un asesoramiento genético integral a la familia.
Discusión
La literatura actual sobre el síndrome de duplicación 15q11.2–q13.3 (Dup15q) destaca la importancia de un diagnóstico genético temprano y preciso debido a su amplio espectro fenotípico y a la superposición con otros trastornos del neurodesarrollo, como los síndromes de Angelman y Prader–Willi. En el caso descrito, múltiples características clínicas —incluyendo retraso global del desarrollo, hipotonía, epilepsia refractaria, trastorno del espectro autista y rasgos dismórficos leves— motivaron una evaluación genética exhaustiva que condujo a la confirmación del diagnóstico mediante array-CGH.
Este enfoque diagnóstico es consistente con los hallazgos de Bisba et al., quienes recomiendan el microarreglo cromosómico (CMA) como prueba de primera línea para detectar variaciones en el número de copias en pacientes con fenotipos complejos. En su estudio, más del 50 % de los individuos diagnosticados con Dup15q presentaron discapacidad intelectual, alteraciones del lenguaje y rasgos autistas, todos ellos también observados en este paciente [1].
De manera similar, Rabeya Akter Mim et al., en una cohorte de 260 niños con trastornos del neurodesarrollo, encontraron que el 3 % presentaba duplicaciones patogénicas en la región 15q11–q13. Estos casos exhibieron perfiles clínicos comparables, reforzando el valor diagnóstico de las pruebas genéticas no solo como herramienta diagnóstica, sino también como guía para el manejo clínico y el asesoramiento familiar [14].
Uno de los aspectos más desafiantes de este caso fue la epilepsia refractaria, un hallazgo frecuente en el síndrome Dup15q. Estudios como el de Elamin et al. (2023) han propuesto una base molecular para esta manifestación, demostrando alteraciones en la inactivación de los canales de sodio en modelos celulares derivados de pacientes. Esta evidencia respalda el desarrollo de terapias dirigidas, que podrían ofrecer alternativas de tratamiento más eficaces en el futuro [2].
En relación con el diagnóstico genético, diversos autores han abogado por la integración sistemática del microarreglo cromosómico (CMA) en la evaluación inicial de pacientes con autismo, discapacidad intelectual y epilepsia [6,10]. En este paciente, el array-CGH estableció con precisión la etiología subyacente, confirmando su utilidad clínica como herramienta diagnóstica de alta resolución. Su capacidad para detectar duplicaciones submicroscópicas con alta sensibilidad permitió identificar de manera precisa la alteración estructural responsable del fenotipo clínico, consolidando su papel como piedra angular de la genética médica moderna.
Asimismo, Bisba et al. enfatizaron la importancia del análisis de metilación para caracterizar las duplicaciones en la región 15q11–q13, particularmente para determinar el origen parental. Este factor es relevante desde el punto de vista pronóstico, ya que las duplicaciones maternas se asocian con fenotipos clínicos más severos. Aunque esta información no estuvo disponible en el presente caso, sigue siendo esencial para el asesoramiento genético y la planificación familiar [1].
En resumen, este caso clínico ilustra cómo una evaluación clínica detallada, complementada con herramientas diagnósticas moleculares como el array-CGH, permite la identificación precisa de una causa genética subyacente y facilita un manejo integral del paciente. La experiencia descrita es concordante con la literatura actual y subraya la necesidad de promover el acceso a estudios de citogenética molecular en entornos clínicos con alta sospecha de anomalías genómicas.
Conclusión
El síndrome de duplicación 15q11.2–q13.3 representa un desafío diagnóstico en pediatría debido a su heterogeneidad clínica y a la superposición fenotípica con otros trastornos del neurodesarrollo. En el presente caso, la presencia de epilepsia refractaria, trastorno del espectro autista y discapacidad intelectual motivó la búsqueda de una causa genética subyacente, la cual fue confirmada mediante microarreglo cromosómico (array-CGH).
La aplicación de esta técnica permitió identificar una duplicación submicroscópica patogénica en la región 15q11.2–q13.3, estableciendo así el diagnóstico etiológico y facilitando un abordaje clínico más dirigido. El array-CGH demostró ser un método de alta resolución con un valor diagnóstico significativo en pacientes con fenotipos neurológicos complejos, ya que permite la detección precisa de variaciones en el número de copias que no son identificables mediante métodos convencionales.
Este caso pone de relieve la necesidad de integrar los estudios de citogenética molecular en la evaluación inicial de pacientes con alta sospecha clínica, así como la importancia del diagnóstico temprano para orientar las decisiones terapéuticas, anticipar el pronóstico y ofrecer un asesoramiento genético adecuado a las familias. La medicina personalizada, basada en la comprensión molecular de la enfermedad, se está consolidando como un pilar fundamental en el manejo de los trastornos del neurodesarrollo.
Referencias
[1] Bisba M, Malamaki C, Constantoulakis P, Vittas S. Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and 14 New Cases. Genes (Basel). 2024 Oct 1;15(10).
[2] Elamin M, Lemtiri-Chlieh F, Robinson TM, Levine ES. Dysfunctional sodium channel kinetics as a novel epilepsy mechanism in chromosome 15q11-q13 duplication syndrome. Epilepsia. 2023 Sep 1;64(9):2515–27.
[3] Piard J, Philippe C, Marvier M, Beneteau C, Roth V, Valduga M, et al. Clinical and molecular characterization of a large family with an interstitial 15q11q13 duplication. Am J Med Genet A. 2010 Aug;152(8):1933–41.
[4] Shehi E, Shah H, Singh A, Pampana VS, Kaur G. The Linkage Between Autism Spectrum Disorder and Dup15q Syndrome: A Case Report. Cureus. 2022 Apr 17;
[5] Urraca N, Cleary J, Brewer V, Pivnick EK, Mcvicar K, Thibert RL, et al. The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Research. 2013 Aug;6(4):268–79.
[6] Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine. 2021 Nov 1;23(11):2029–37.
[7] Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am J Hum Genet. 2010 May 14;86(5):749–64.
[8] Cucinotta F, Lintas C, Tomaiuolo P, Baccarin M, Picinelli C, Castronovo P, et al. Diagnostic yield and clinical impact of chromosomal microarray analysis in autism spectrum disorder. Mol Genet Genomic Med. 2023 Aug 1;11(8).
[9] Gürkan H, Atli Eİ, Atli E, Bozatli L, Araz Altay M, Yalçintepe S, et al. Chromosomal microarray analysis in Turkish patients with unexplained developmental delay and intellectual developmental disorders. Noropsikiyatri Arsivi. 2020;57(3):177–91.
[10] Jang W, Kim Y, Han E, Park J, Chae H, Kwon A, et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: A prospective multicenter study in korea. Ann Lab Med. 2019;39(3):299–310.
[11] Aldosari AN, Aldosari TS. Comprehensive evaluation of the child with global developmental delays or intellectual disability. Vol. 67, Clinical and Experimental Pediatrics. Korean Pediatric Society; 2024. p. 435–46.
[12] Ma Y, Yang R, Yan X, Song X, Zhan F. Genetic analysis and prenatal diagnosis of 15q11-q13 microduplication syndrome. Journal of Maternal-Fetal and Neonatal Medicine. 2025;38(1).
[13] Jeste S, DiStefano C. Can Preclinical Insights Give Us Hope for Effective Treatments for Epilepsy in 15q11-q13 Duplication Syndrome? Vol. 90, Biological Psychiatry. Elsevier Inc.; 2021. p. 735–7.
[14] Mim RA, Soorajkumar A, Kosaji N, Rahman MM, Sarker S, Karuvantevida N, et al. Expanding deep phenotypic spectrum associated with atypical pathogenic structural variations overlapping 15q11–q13 imprinting region. Brain Behav. 2024 Apr 1;14(4).
